Voronota-JS is an expansion of the core Voronota software. Voronota-JS provides a way to write JavaScript scripts for the comprehensive analysis of macromolecular structures, including the Voronoi tesselation-based analysis.
Currently, the Voronota-JS package contains several executables:
Voronota-JS relies on several externally developed software projects (big thanks to their authors):
Please refer to the core Voronota quick install guide.
Download the latest archive from the official downloads page: https://github.com/kliment-olechnovic/voronota/releases.
The archive contains the Voronota-JS software in the ‘expansion_js’ subdirectory. There is a ready-to-use statically compiled ‘voronota-js’ program for 64 bit Linux systems. This executable can be rebuilt from the provided source code to work on any modern Linux, macOS or Windows operating systems.
Voronota-JS has no required external dependencies, only a C++14-compliant compiler is needed to build it.
You can build using CMake for makefile generation. Starting in the directory containing “CMakeLists.txt” file, run the sequence of commands:
cmake ./
makeAlternatively, to keep files more organized, CMake can be run in a separate “build” directory:
mkdir build
cd build
cmake ../
make
cp ./voronota-js ../voronota-jsFor example, “voronota-js” executable can be built from the sources in “src” directory using GNU C++ compiler:
g++ -std=c++14 -I"./src/dependencies" -O3 -o "./voronota-js" $(find ./src/ -name '*.cpp')Two utility scripts for generating Voronoi tessellation-based data graphs are included in the Voronota-JS expansion of Voronota:
voronota-js-receptor-data-graph - for generating graphs with an extended set of annotationsvoronota-js-lt-data-graph - for generating basic graphs, but much faster, using the Voronota-LT algorithmThese scripts are available in the latest version of the Voronota package, in the expanson_js subdirectory.
The command-line interface of the voronota-js-receptor-data-graph script is described below:
'voronota-js-receptor-data-graph' script describes a receptor protein structure as an annotated graph.
Options:
--input string * path to input protein file
--probe-min number scanning probe radius minimum, default is 2.0
--probe-max number scanning probe radius maximum, default is 30.0
--buriedness-core number buriedness minimum for pocket start, default is 0.7
--buriedness-rim number buriedness maximum for pocket end, default is 0.4
--subpockets number number of sorted subpockets to include, default is 999999
--output-dir string * output directory path
--output-naming string output files naming mode, default is 'BASENAME/name', other possibilities are 'BASENAME_name' and 'BASENAME/BASENAME_name'
--help | -h flag to display help message and exit
Standard output:
Information messages in stdout, error messages in stderr
Examples:
voronota-js-receptor-data-graph --input "./2zsk.pdb" --output-dir "./output"
voronota-js-receptor-data-graph --input "./2zsk.pdb" --output-dir "./output" --probe-min 1 --probe-max 5 --buriedness-core 0.8 --buriedness-rim 0.7 --subpockets 1Running
voronota-js-receptor-data-graph --input "./2zsk.pdb" --output-dir "./output"generates four .TSV files:
./output/2zsk/atom_graph_nodes.tsv - atom-level graph nodes table file, one row per node, every node represents an atom./output/2zsk/atom_graph_links.tsv - atom-level graph links (edges) table file, one row per link, every link represents an atom-atom contact./output/2zsk/residue_graph_nodes.tsv - residue-level graph nodes table file, one row per node, every node represents a residue./output/2zsk/residue_graph_links.tsv - residue-level graph links (edges) table file, one row per link, every link represents a residue-residue contactIt also generates .PDB files with “buriedness” and “pocketness” values written as b-factors - in case a user wants to visualize those values in a 3D viewer.
Example (first 10 lines) from the file ./output/2zsk/atom_graph_nodes.tsv:
ID_chainID ID_resSeq ID_iCode ID_serial ID_altLoc ID_resName ID_name atom_index residue_index atom_type residue_type center_x center_y center_z radius sas_area solvdir_x solvdir_y solvdir_z voromqa_sas_energy voromqa_depth voromqa_score_a voromqa_score_r volume volume_vdw ev14 ev28 ev56 ufsr_a1 ufsr_a2 ufsr_a3 ufsr_b1 ufsr_b2 ufsr_b3 ufsr_c1 ufsr_c2 ufsr_c3 buriedness pocketness
A 1 . . . MET N 0 0 97 12 22.493 56.29 12.4 1.7 53.351 0.559085 0.82758 -0.0503525 57.9168 1 1.01688e-07 0.167578 66.5845 15.242 0.125304 0.124587 0.121036 30.0876 112.815 -196.313 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.132536 0
A 1 . . . MET CA 1 0 93 12 21.658 55.076 12.14 1.9 4.72457 0.135243 0.432065 -0.891644 6.17629 1 0.015941 0.167578 26.134 13.3463 0.0448295 0.039199 0.0265035 30.0876 112.815 -196.313 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.0847213 0
A 1 . . . MET C 2 0 92 12 20.271 55.177 12.793 1.75 2.62013 -0.250994 0.910664 -0.328166 5.18333 1 0.292879 0.167578 14.0446 8.87448 0.601112 0.563979 0.457316 30.0876 112.815 -196.313 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.13229 0
A 1 . . . MET O 3 0 98 12 20.135 55.689 13.912 1.49 11.2317 0.372405 0.737202 0.56378 8.18408 1 0.361824 0.167578 31.8395 9.38393 0.661347 0.635732 2 30.0876 112.815 -196.313 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.139913 0
A 1 . . . MET CB 4 0 94 12 22.393 53.825 12.646 1.91 27.6946 0.886864 -0.0929711 0.452581 17.787 1 0.167248 0.167578 74.006 23.068 0.63118 0.590042 0.584411 30.0876 112.815 -196.313 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.121678 0
A 2 . . . LYS N 5 1 89 11 19.254 54.687 12.079 1.7 2.52782 -0.0429483 0.809307 -0.585814 3.47919 1 0.052448 0.472209 17.3995 10.0202 0.55985 0.535622 0.457316 29.1886 119.733 -71.1108 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.0643745 0
A 2 . . . LYS CA 6 1 84 11 17.871 54.701 12.555 1.9 2.06927 0.0828678 0.93132 -0.354648 1.32819 1 0.342294 0.472209 18.5844 12.7854 0.62449 0.611648 0.645691 29.1886 119.733 -71.1108 41.8069 256.541 -1727.84 37.0975 249.607 439.762 0.0846022 0
A 2 . . . LYS C 7 1 83 11 17.712 53.714 13.699 1.75 0.033581 0.809845 -0.165563 0.562797 0.0571512 1 0.698036 0.472209 11.1993 8.68042 0.965086 2 2 29.1886 119.733 -71.1108 41.8069 256.541 -1727.84 37.0975 249.607 439.762 2 0
A 2 . . . LYS O 8 1 91 11 18.114 52.551 13.6 1.49 1.14708 0.796577 -0.0907621 0.597685 0.209446 1 0.571392 0.472209 21.023 9.3442 0.965086 2 2 29.1886 119.733 -71.1108 41.8069 256.541 -1727.84 37.0975 249.607 439.762 2 0Description of the columns:
Example (first 10 lines) from the file ./output/2zsk/atom_graph_links.tsv:
ID1_chainID ID1_resSeq ID1_iCode ID1_serial ID1_altLoc ID1_resName ID1_name ID2_chainID ID2_resSeq ID2_iCode ID2_serial ID2_altLoc ID2_resName ID2_name atom_index1 atom_index2 area boundary distance voromqa_energy seq_sep_class covalent_bond
A 1 . . . MET N A 1 . . . MET CA 0 1 15.4593 6.06454 1.4962 0 0 1
A 1 . . . MET N A 1 . . . MET C 0 2 2.49966 1.78477 2.51605 0 0 0
A 1 . . . MET N A 1 . . . MET O 0 3 6.83024 5.45489 2.86488 0 0 0
A 1 . . . MET N A 1 . . . MET CB 0 4 9.44152 6.97601 2.47926 0 0 0
A 1 . . . MET CA A 1 . . . MET C 1 2 8.27879 0.824826 1.53635 0 0 1
A 1 . . . MET CA A 1 . . . MET O 1 3 0.751241 0 2.41563 0 0 0
A 1 . . . MET CA A 1 . . . MET CB 1 4 12.4635 1.04702 1.53664 0 0 1
A 1 . . . MET CA A 2 . . . LYS N 1 5 4.95624 1.021 2.43603 0 1 0
A 1 . . . MET CA A 224 . . . ALA O 1 1783 2.2368 0 3.58016 -0.886374 5 0Description of the columns:
Example (first 10 lines) from the file ./output/2zsk/residue_graph_nodes.tsv:
ID_chainID ID_resSeq ID_iCode ID_serial ID_altLoc ID_resName ID_name residue_index residue_type residue_sas_area residue_volume residue_voromqa_sas_energy residue_mean_voromqa_depth voromqa_score_r residue_min_ev14 residue_max_ev14 residue_mean_ev14 residue_min_ev28 residue_max_ev28 residue_mean_ev28 residue_min_ev56 residue_max_ev56 residue_mean_ev56 residue_min_buriedness residue_max_buriedness residue_mean_buriedness
A 1 . . . MET . 0 12 99.622 212.609 95.2474 1 0.167578 0.0448295 0.661347 0.335069 0.039199 0.635732 0.319117 0.0265035 0.584411 0.240568 0.0847213 0.139913 0.128075
A 2 . . . LYS . 1 11 26.6093 132.673 2.76791 1 0.472209 0.502281 0.965086 0.537789 0.498853 0.611648 0.488983 0.457316 0.645691 0.452329 0.0643745 0.0846022 0.0788722
A 3 . . . LYS . 2 11 66.3108 253.264 -53.6353 1.44444 0.613378 0.337858 0.965086 0.366175 0.337066 0.621141 0.36095 0.334376 0.473626 0.351902 0.337636 0.34682 0.337817
A 4 . . . ILE . 3 9 0 167.223 0 2.5 0.643901 2 2 2 2 2 2 2 2 2 2 2 2
A 5 . . . GLY . 4 7 0 63.6288 0 2.25 0.996122 2 2 2 2 2 2 2 2 2 2 2 2
A 6 . . . ILE . 5 9 1.9557 175.105 1.55475 1.75 0.913145 0.978935 0.994222 0.981069 2 2 2 2 2 2 2 2 2
A 7 . . . ILE . 6 9 0.997957 170.309 0.912408 2 0.895152 0.990166 0.990742 0.990307 2 2 2 2 2 2 2 2 2
A 8 . . . GLY . 7 7 0 69.5537 0 2 0.970268 2 2 2 2 2 2 2 2 2 2 2 2
A 9 . . . GLY . 8 7 0 63.8229 0 2 0.848999 2 2 2 2 2 2 2 2 2 2 2 2Description of the columns: they are per-residue summaries of the columns from the atom graph nodes file.
Example (first 10 lines) from the file ./output/2zsk/residue_graph_links.tsv:
ID1_chainID ID1_resSeq ID1_iCode ID1_serial ID1_altLoc ID1_resName ID1_name ID2_chainID ID2_resSeq ID2_iCode ID2_serial ID2_altLoc ID2_resName ID2_name area boundary distance voromqa_energy
A 1 . . . MET . A 2 . . . LYS . 26.7853 5.60234 1.33573 0
A 1 . . . MET . A 3 . . . LYS . 11.6844 4.40933 3.46409 4.30572
A 1 . . . MET . A 34 . . . PHE . 7.33832 7.65326 5.72583 -12.2026
A 1 . . . MET . A 36 . . . PRO . 10.5094 4.20942 5.41294 -4.09459
A 1 . . . MET . A 37 . . . GLU . 2.13388 6.22514 5.51268 2.25328
A 1 . . . MET . A 75 . . . GLU . 4.96855 3.02668 3.82408 6.90425
A 1 . . . MET . A 224 . . . ALA . 9.9518 0 3.34954 -0.823021
A 1 . . . MET . A 225 . . . SER . 10.3855 6.90767 4.80928 3.00817
A 1 . . . MET . A 226 . . . GLU . 6.02953 3.85763 4.80279 -0.209601Description of the columns: they are per-residue-residue contact summaries of the columns from the atom graph links file.
The command-line interface of the voronota-js-lt-data-graph script is described below:
'voronota-js-lt-graph' script describes a molecular structure as a graph.
Options:
--input string * path to input protein file
--probe number probe radius is 1.4
--output-dir string * output directory path
--output-naming string output files naming mode, default is 'BASENAME/name', other possibilities are 'BASENAME_name' and 'BASENAME/BASENAME_name'
--help | -h flag to display help message and exit
Standard output:
Information messages in stdout, error messages in stderr
Examples:
voronota-js-lt-graph --input "./2zsk.pdb" --output-dir "./output"
voronota-js-lt-graph --input "./2zsk.pdb" --output-dir "./output" --probe 1.4Running
voronota-js-lt-data-graph --input "./2zsk.pdb" --output-dir "./output"generates four .TSV files:
./output/2zsk/atom_graph_nodes.tsv - atom-level graph nodes table file, one row per node, every node represents an atom./output/2zsk/atom_graph_links.tsv - atom-level graph links (edges) table file, one row per link, every link represents an atom-atom contact./output/2zsk/residue_graph_nodes.tsv - residue-level graph nodes table file, one row per node, every node represents a residue./output/2zsk/residue_graph_links.tsv - residue-level graph links (edges) table file, one row per link, every link represents a residue-residue contactExample (first 10 lines) from the file ./output/2zsk/atom_graph_nodes.tsv:
ID_chainID ID_resSeq ID_iCode ID_serial ID_altLoc ID_resName ID_name atom_index residue_index atom_type residue_type center_x center_y center_z radius sas_area volume
A 1 . . . MET N 0 0 97 12 22.493 56.29 12.4 1.7 53.3426 63.8989
A 1 . . . MET CA 1 0 93 12 21.658 55.076 12.14 1.9 4.70418 30.7944
A 1 . . . MET C 2 0 92 12 20.271 55.177 12.793 1.75 2.62416 15.6203
A 1 . . . MET O 3 0 98 12 20.135 55.689 13.912 1.49 11.1833 27.3368
A 1 . . . MET CB 4 0 94 12 22.393 53.825 12.646 1.91 27.5103 75.9687
A 2 . . . LYS N 5 1 89 11 19.254 54.687 12.079 1.7 2.52533 14.778
A 2 . . . LYS CA 6 1 84 11 17.871 54.701 12.555 1.9 2.06606 23.2376
A 2 . . . LYS C 7 1 83 11 17.712 53.714 13.699 1.75 0.0340018 12.7911
A 2 . . . LYS O 8 1 91 11 18.114 52.551 13.6 1.49 1.12595 15.7616Description of the columns:
Example (first 10 lines) from the file ./output/2zsk/atom_graph_links.tsv:
ID1_chainID ID1_resSeq ID1_iCode ID1_serial ID1_altLoc ID1_resName ID1_name ID2_chainID ID2_resSeq ID2_iCode ID2_serial ID2_altLoc ID2_resName ID2_name atom_index1 atom_index2 area boundary distance
A 1 . . . MET N A 1 . . . MET CA 0 1 16.5137 6.06483 1.4962
A 1 . . . MET N A 1 . . . MET CB 0 4 8.78012 6.97738 2.47926
A 1 . . . MET N A 1 . . . MET C 0 2 2.26495 1.78545 2.51605
A 1 . . . MET N A 1 . . . MET O 0 3 5.95355 5.4563 2.86488
A 1 . . . MET CA A 1 . . . MET CB 1 4 14.3565 1.04322 1.53664
A 1 . . . MET CA A 1 . . . MET C 1 2 9.42943 0.82315 1.53635
A 1 . . . MET CA A 2 . . . LYS N 1 5 5.50647 1.01891 2.43603
A 1 . . . MET CA A 224 . . . ALA O 1 1783 2.13187 0 3.58016
A 1 . . . MET CA A 226 . . . GLU OXT 1 1800 5.78086 3.06435 4.80279Description of the columns:
Example (first 10 lines) from the file ./output/2zsk/residue_graph_nodes.tsv:
ID_chainID ID_resSeq ID_iCode ID_serial ID_altLoc ID_resName ID_name residue_type residue_sas_area residue_volume
A 1 . . . MET . 12 99.3646 213.619
A 2 . . . LYS . 11 26.5311 132.432
A 3 . . . LYS . 11 66.24 256.468
A 4 . . . ILE . 9 0 168.399
A 5 . . . GLY . 7 0 62.1498
A 6 . . . ILE . 9 1.93343 175.439
A 7 . . . ILE . 9 0.989463 171.733
A 8 . . . GLY . 7 0 69.6737
A 9 . . . GLY . 7 0 63.6192Description of the columns: they are per-residue summaries of the columns from the atom graph nodes file.
Example (first 10 lines) from the file ./output/2zsk/residue_graph_links.tsv:
ID1_chainID ID1_resSeq ID1_iCode ID1_serial ID1_altLoc ID1_resName ID1_name ID2_chainID ID2_resSeq ID2_iCode ID2_serial ID2_altLoc ID2_resName ID2_name area boundary distance
A 1 . . . MET . A 2 . . . LYS . 27.3555 5.59216 1.33573
A 1 . . . MET . A 3 . . . LYS . 11.287 4.39984 3.46409
A 1 . . . MET . A 34 . . . PHE . 7.35286 7.65587 5.72583
A 1 . . . MET . A 36 . . . PRO . 10.5861 4.18204 5.41294
A 1 . . . MET . A 37 . . . GLU . 2.13882 6.27953 5.51268
A 1 . . . MET . A 75 . . . GLU . 4.52381 3.02635 3.82408
A 1 . . . MET . A 224 . . . ALA . 9.38126 0 3.34954
A 1 . . . MET . A 225 . . . SER . 11.0272 6.90226 4.80928
A 1 . . . MET . A 226 . . . GLU . 6.14355 3.85731 4.80279Description of the columns: they are per-residue-residue contact summaries of the columns from the atom graph links file.
The script ‘voronota-js-ligand-cadscore’ for the protein-ligand variation of CAD-score is included to the Voronota-JS expansion of Voronota.
The ‘voronota-js-ligand-cadscore’ scripts has a special input mode for CASP15 protein-ligand model files: when using –casp15-* options for specifying input, the input files are split into receptor and ligand parts and passed to the same script with basic input options. The target input in the CASP15 mode should be formatted in the same as the model input. The basic input mode option is more flexible, but it requires all the receptor and ligand files available separately, and the ligand IDs (names) to be specified explicitly.
For example, let us take two files with protein-ligand models from CASP15: ‘H1135LG035_1’ and ‘H1135LG046_1’. Then let us execute the following command:
voronota-js-ligand-cadscore \
--casp15-target ./T1118v1LG035_1 \
--casp15-model ./T1118v1LG046_1 \
--casp15-target-pose 1 \
--casp15-model-pose 2 \
| column -twhich outputs:
target model contacts_set CAD_score target_area model_area
T1118v1LG035_1_pose1 T1118v1LG046_1_pose2 interface 0.568301 648.036 808.139
T1118v1LG035_1_pose1 T1118v1LG046_1_pose2 interface_plus_adjacent 0.629109 1781.9 2189.88Note, that CAD-score values will be different if we swap target and model inputs. The script can do the swap automatically and output additional results when the ‘–and-swap true’ is added to the arguments, then the output will look as follows:
target model contacts_set CAD_score target_area model_area
T1118v1LG035_1_pose1 T1118v1LG046_1_pose2 interface 0.568301 648.036 808.139
T1118v1LG035_1_pose1 T1118v1LG046_1_pose2 interface_plus_adjacent 0.629109 1781.9 2189.88
T1118v1LG046_1_pose2 T1118v1LG035_1_pose1 interface 0.528076 808.139 648.036
T1118v1LG046_1_pose2 T1118v1LG035_1_pose1 interface_plus_adjacent 0.519405 2189.88 1781.9The output table columns mean the following
There are some important aspects:
Examples of files generated using the ‘–details-dir’ option:
Example pymol call for displaying files generated using the ‘–drawing-dir’ option:
pymol \
draw_T1118v1LG035_1_pose1_interface_and_adjacent_contacts_in_pymol.py \
T1118v1LG035_1_pose1_cutout_interface_residues_ligand.pdb \
T1118v1LG035_1_pose1_cutout_interface_residues_receptor.pdbExample of visualized contacts (with direct interface contacts in green, adjacent contacts in yellow, ligand atoms in red, receptor atoms in cyan): protein_ligand_3D.png.
‘voronota-js-voromqa’ script provides an interface to VoroMQA dark (newer) and light (classic) methods.
Options:
--input | -i string * input file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--sequence-file string input sequence file
--select-contacts string query to select contacts, default is '[-min-seq-sep 2]'
--inter-chain string set query to select contacts to '[-inter-chain]'
--constraints-file string input distance constraints file
--output-table-file string output table file path, default is '_stdout' to print to stdout
--output-dark-scores string output PDB file with dark scores as B-factors
--output-light-scores string output PDB file with light scores as B-factors
--output-alignment string output alignment sequence alignment file
--cache-dir string path to cache directory
--tour-sort string tournament sorting mode, default is '_mono', options are '_homo', '_hetero' or custom
--sbatch-parameters string parameters to run Slurm sbatch if input is '_list'
--split-models-prefix string name prefix for splitting input PDB file by models
--smoothing-window number residue scores smoothing window size, default is 0
--processors number maximum number of processors to use, default is 1
--casp-b-factors flag to write CASP-required B-factors in output PDB files
--use-scwrl flag to use Scwrl4 to rebuild side-chains
--guess-chain-names flag to guess chain names based on sequence nubmering
--order-by-residue-id flag to order atoms by residue ids
--input-is-script flag to treat input file as vs script
--as-assembly flag to treat input file as biological assembly
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-voromqa --input model.pdb
voronota-js-voromqa --cache-dir ./cache --input model.pdb
ls *.pdb | voronota-js-voromqa --cache-dir ./cache --input _list
ls *.pdb | voronota-js-voromqa --cache-dir ./cache --input _list | column -t
ls *.pdb | voronota-js-voromqa --cache-dir ./cache --input _list \
--processors 8 \
--inter-chain \
--tour-sort _hetero
ls *.pdb | voronota-js-voromqa --cache-dir ./cache --input _list \
--processors 8 \
--select-contacts '[-a1 [-chain A -rnum 1:500] -a2 [-chain B -rnum 1:500]]' \
--tour-sort '-columns full_dark_score sel_energy -multipliers 1 -1 -tolerances 0.02 0.0'‘voronota-js-only-global-voromqa’ script computes global VoroMQA scores and can use fast caching.
Options:
--input | -i string * input file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--output-table-file string output table file path, default is '_stdout' to print to stdout
--output-dark-pdb string output path for PDB file with VoroMQA-dark scores, default is ''
--output-light-pdb string output path for PDB file with VoroMQA-light scores, default is ''
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--cache-dir string cache directory path to store results of past calls
--run-faspr string path to FASPR binary to rebuild side-chains
--input-is-script flag to treat input file as vs script
--as-assembly flag to treat input file as biological assembly
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-only-global-voromqa --input model.pdb
ls *.pdb | voronota-js-only-global-voromqa --input _list --processors 8 | column -t‘voronota-js-membrane-voromqa’ script provides an interface to the VoroMQA-based method for assessing membrane protein structures.
Options:
--input | -i string * input file path
--restrict-input string query to restrict input atoms, default is '[]'
--membrane-width number membrane width or list of widths to use, default is 25.0
--output-local-scores string prefix to output PDB files with local scores as B-factors
--as-assembly flag to treat input file as biological assembly
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-membrane-voromqa --input model.pdb --membrane-width 30.0
voronota-js-membrane-voromqa --input model.pdb --membrane-width 20.0,25.0,30.0
voronota-js-membrane-voromqa --input model.pdb \
--membrane-width 20,25,30 \
--output-local-scores ./local_scores/‘voronota-js-ifeatures-voromqa’ script computes multiple VoroMQA-based features of protein-protein complexes.
Options:
--input | -i string * input file path or '_list' to read file paths from stdin
--output-table-file string output table file path, default is '_stdout' to print to stdout
--processors number maximum number of processors to use, default is 1
--use-scwrl flag to use Scwrl4 to rebuild side-chains
--as-assembly flag to treat input file as biological assembly
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-ifeatures-voromqa --input model.pdb
ls *.pdb | voronota-js-ifeatures-voromqa --input _list --processors 8 | column -t‘voronota-js-fast-iface-voromqa’ script rapidly computes VoroMQA-based interface energy of protein complexes.
Options:
--input | -i string * input file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect inter-chain contacts, default is '[]'
--constraints-required string query to check required contacts, default is ''
--constraints-banned string query to check banned contacts, default is ''
--constraint-clashes number max allowed clash score, default is 0.9
--output-table-file string output table file path, default is '_stdout' to print to stdout
--output-ia-contacts-file string output inter-atom contacts file path, default is ''
--output-ir-contacts-file string output inter-residue contacts file path, default is ''
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--run-faspr string path to FASPR binary to rebuild side-chains
--input-is-script flag to treat input file as vs script
--as-assembly flag to treat input file as biological assembly
--detailed-times flag to output detailed times
--score-symmetry flag to score interface symmetry
--blanket flag to keep nucleic acids and use blanket potential
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-fast-iface-voromqa --input model.pdb
ls *.pdb | voronota-js-fast-iface-voromqa --input _list --processors 8 | column -t‘voronota-js-fast-iface-cadscore’ script rapidly computes interface CAD-score for two protein complexes.
Options:
--target | -t string * target file path
--model | -m string * model file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect inter-chain contacts, default is '[]'
--subselect-site string query to subselect atoms for binding site scoring, default is '[]'
--output-table-file string output table file path, default is '_stdout' to print to stdout
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--run-faspr string path to FASPR binary to rebuild side-chains
--permuting-allowance number maximum number of chains for exhaustive remapping, default is 4
--as-assembly flag to treat input files as biological assemblies
--include-heteroatoms flag to not discard heteroatoms
--remap-chains flag to calculate and use optimal chains remapping
--remap-chains-logging flag to print log of chains remapping to stderr
--ignore-residue-names flag to ignore residue names in residue identifiers
--test-common-ids flag to fail quickly if there are no common residues
--crude flag to enable very crude faster mode
--lt flag to enable voronota-lt faster mode
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-fast-iface-cadscore --target target.pdb --model model.pdb
ls *.pdb | voronota-js-fast-iface-cadscore --target target.pdb --model _list --processors 8 | column -t‘voronota-js-fast-iface-cadscore-matrix’ script rapidly computes interface CAD-score between complexes.
Options:
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect inter-chain contacts, default is '[]'
--output-table-file string output table file path, default is '_stdout' to print to stdout
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--permuting-allowance number maximum number of chains for exhaustive remapping, default is 4
--as-assembly flag to treat input files as biological assemblies
--include-heteroatoms flag to not discard heteroatoms
--remap-chains flag to calculate and use optimal chains remapping
--ignore-residue-names flag to ignore residue names in residue identifiers
--crude flag to enable very crude faster mode
--lt flag to enable voronota-lt faster mode
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
ls *.pdb | voronota-js-fast-iface-cadscore-matrix | column -t
find ./complexes/ -type f -name '*.pdb' | voronota-js-fast-iface-cadscore-matrix > "full_matrix.txt"
(find ./group1/ -type f | awk '{print $1 " a"}' ; find ./group2/ -type f | awk '{print $1 " b"}') | voronota-js-fast-iface-cadscore-matrix > "itergroup_matrix.txt"‘voronota-js-fast-iface-contacts’ script rapidly computes contacts of inter-chain interface in a molecular complex.
Options:
--input string * input file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect inter-chain contacts, default is '[]'
--output-contacts-file string output table file path, default is '_stdout' to print to stdout
--output-bsite-file string output binding site table file path, default is ''
--output-drawing-script string output PyMol drawing script file path, default is ''
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--run-faspr string path to FASPR binary to rebuild side-chains
--custom-radii-file string path to file with van der Waals radii assignment rules
--with-sas-areas flag to also compute and output solvent-accessible areas of interface residue atoms
--coarse-grained flag to output inter-residue contacts
--input-is-script flag to treat input file as vs script
--as-assembly flag to treat input file as biological assembly
--use-hbplus flag to run 'hbplus' to tag H-bonds
--expand-ids flag to output expanded IDs
--og-pipeable flag to format output to be pipeable to 'voronota query-contacts'
--help | -h flag to display help message and exit
Standard output:
tab-separated table of contacts
Examples:
voronota-js-fast-iface-contacts --input "./model.pdb" --expand-ids > "./contacts.tsv"
voronota-js-fast-iface-contacts --input "./model.pdb" --with-sas-areas --coarse-grained --og-pipeable | voronota query-contacts --summarize-by-first
cat "./model.pdb" | voronota-js-fast-iface-contacts --input _stream --with-sas-areas --coarse-grained --og-pipeable | voronota query-contacts --summarize
ls *.pdb | voronota-js-fast-iface-contacts --input _list --processors 8 --output-contacts-file "./output/-BASENAME-.tsv"‘voronota-js-fast-iface-data-graph’ script generates interface data graphs of protein complexes.
Options:
--input string * input file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect inter-chain contacts, default is '[]'
--with-reference string input reference complex structure file path, default is ''
--output-data-prefix string * output data files prefix
--output-table-file string output table file path, default is '_stdout' to print to stdout
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--run-faspr string path to FASPR binary to rebuild side-chains
--coarse-grained flag to output a coarse-grained graph
--input-is-script flag to treat input file as vs script
--as-assembly flag to treat input file as biological assembly
--help | -h flag to display help message and exit
Standard output:
space-separated table of generated file paths
Examples:
voronota-js-fast-iface-data-graph --input model.pdb --output-prefix ./data_graphs/
ls *.pdb | voronota-js-fast-iface-data-graph --input _list --processors 8 --output-prefix ./data_graphs/ | column -t‘voronota-js-voroif-gnn’ scores protein-protein interfaces using the VoroIF-GNN method
Options:
--input string * input file path, or '_list' to read multiple input files paths from stdin
--gnn string GNN package file or directory with package files, default is '${SCRIPT_DIRECTORY}/voroif/gnn_packages/v1'
--gnn-add string additional GNN package file or directory with package files, default is ''
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect inter-chain contacts, default is '[]'
--as-assembly string flag to treat input file as biological assembly
--input-is-script string flag to treat input file as vs script
--conda-path string conda installation path, default is ''
--conda-env string conda environment name, default is ''
--faspr-path string path to FASPR binary, default is ''
--run-faspr string flag to rebuild sidechains using FASPR, default is 'false'
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin, default is '_stream'
--local-column string flag to add per-residue scores to the global output table, default is 'false'
--cache-dir string cache directory path to store results of past calls, default is ''
--output-dir string output directory path for all results, default is ''
--output-pdb-file string output path for PDB file with interface residue scores, default is ''
--output-pdb-mode string mode to write b-factors ('overwrite_all', 'overwrite_iface' or 'combine'), default is 'overwrite_all'
--help | -h flag to display help message and exit
Standard output:
space-separated table of global scores
Important note about output interpretation:
higher GNN scores are better, lower GNN scores are worse (with VoroMQA energy it is the other way around)
Examples:
voronota-js-voroif-gnn --conda-path ~/miniconda3 --input './model.pdb'
voronota-js-voroif-gnn --input './model.pdb' --gnn "${HOME}/git/voronota/expansion_js/voroif/gnn_packages/v1"
find ./models/ -type f | voronota-js-voroif-gnn --conda-path ~/miniconda3 --input _list
Requirements installation example using Miniconda (may need more than 10 GB of disk space):
# prepare Miniconda
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
source ~/miniconda3/bin/activate
# install PyTorch using instructions from 'https://pytorch.org/get-started/locally/'
conda install pytorch torchvision torchaudio pytorch-cuda=12.1 -c pytorch -c nvidia
# install PyTorch Geometric using instructions from 'https://pytorch-geometric.readthedocs.io/en/latest/install/installation.html'
pip install torch_geometric
pip install pyg_lib torch_scatter torch_sparse torch_cluster torch_spline_conv -f https://data.pyg.org/whl/torch-2.1.0+cu121.html
# install Pandas
conda install pandas
# if you do not have R installed in you system, install it (not necessarily using conda, e.g 'sudo apt-get install r-base' in Ubuntu)
conda install r -c conda-forge‘voronota-js-ligand-cadscore’ script computes protein-ligand variation of CAD-score.
Input options, basic:
--target-receptor string * target receptor file path
--target-ligands string * list of target ligand file paths
--target-ligand-ids string * list of target ligand IDs
--model-receptor string * model receptor file path
--model-ligands string * list of model ligand file paths
--model-ligand-ids string * list of model ligand IDs
--target-name string target name to use for output, default is 'target_complex'
--model-name string model name to use for output, default is 'model_complex'
Input options, alternative:
--casp15-target string * target data file in CASP15 format, alternative to --target-* options
--casp15-target-pose string * pose number to select from the target data file in CASP15 format
--casp15-model string * model data file in CASP15 format, alternative to --model-* options
--casp15-model-pose string * pose number to select from the model data file in CASP15 format
Other options:
--table-dir string directory to output scores table file named '${TARGET_NAME}_vs_${MODEL_NAME}.txt'
--details-dir string directory to output lists of contacts used for scoring
--drawing-dir string directory to output files to visualize with pymol
--and-swap string flag to compute everything after swapping target and model, default is 'false'
--ignore-ligand-headers string flag to ignore title header in ligand files
--help | -h display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-ligand-cadscore --casp15-target ./T1118v1LG035_1 --casp15-target-pose 1 --casp15-model ./T1118v1LG046_1 --casp15-model-pose 1
voronota-js-ligand-cadscore \
--target-receptor ./t_protein.pdb --target-ligands './t_ligand1.mol ./t_ligand2.mol ./t_ligand3.mol' --target-ligand-ids 'a a b' \
--model-receptor ./m_protein.pdb --model-ligands './m_ligand1.mol ./m_ligand2.mol ./m_ligand3.mol' --model-ligand-ids 'a a b'‘voronota-js-global-cadscore’ script computes global CAD-score between molecular structures.
Options:
--target | -t string * target file path
--model | -m string * model file path or '_list' to read file paths from stdin
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect contacts, default is '[]'
--output-table-file string output table file path, default is '_stdout' to print to stdout
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--run-faspr string path to FASPR binary to rebuild side-chains
--permuting-allowance number maximum number of chains for exhaustive remapping, default is 4
--as-assembly flag to treat input files as biological assemblies
--include-heteroatoms flag to not discard heteroatoms
--remap-chains flag to calculate and use optimal chains remapping
--remap-chains-logging flag to print log of chains remapping to stderr
--ignore-residue-names flag to ignore residue names in residue identifiers
--test-common-ids flag to fail quickly if there are no common residues
--crude flag to enable very crude faster mode
--lt flag to enable faster mode based on Voronota-LT
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
voronota-js-global-cadscore --target target.pdb --model model.pdb --lt
ls *.pdb | voronota-js-global-cadscore --target target.pdb --model _list --lt --processors 8 | column -t‘voronota-js-global-cadscore-matrix’ script computes global CAD-score for all pairs of provided molecular structures.
Options:
--restrict-input string query to restrict input atoms, default is '[]'
--subselect-contacts string query to subselect contacts, default is '[]'
--output-table-file string output table file path, default is '_stdout' to print to stdout
--processors number maximum number of processors to run in parallel, default is 1
--sbatch-parameters string sbatch parameters to run in parallel, default is ''
--stdin-file string input file path to replace stdin
--permuting-allowance number maximum number of chains for exhaustive remapping, default is 4
--as-assembly flag to treat input files as biological assemblies
--include-heteroatoms flag to not discard heteroatoms
--remap-chains flag to calculate and use optimal chains remapping
--ignore-residue-names flag to ignore residue names in residue identifiers
--crude flag to enable very crude faster mode
--lt flag to enable faster mode based on Voronota-LT
--help | -h flag to display help message and exit
Standard output:
space-separated table of scores
Examples:
ls *.pdb | voronota-js-global-cadscore-matrix --lt | column -t
find ./complexes/ -type f -name '*.pdb' | voronota-js-global-cadscore-matrix --lt > "full_matrix.txt"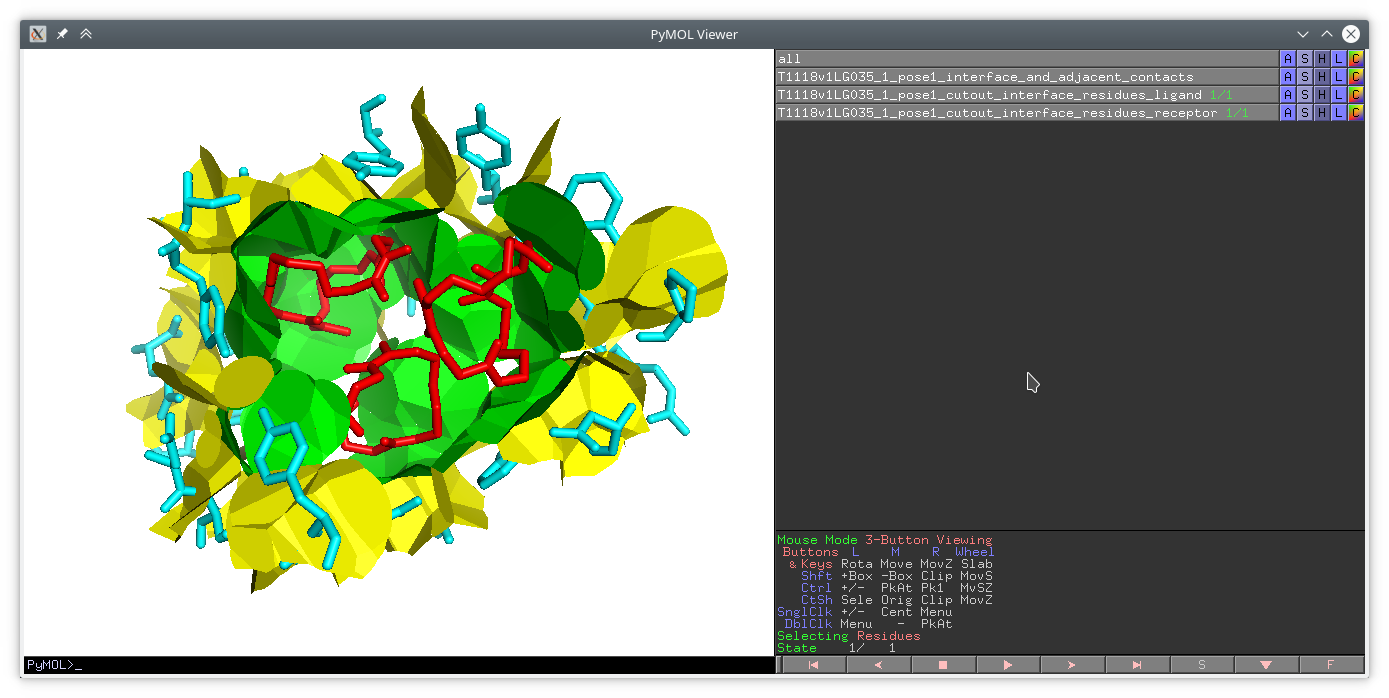{kind=link}